Defining molecular mechanisms of imatinib treatment in GIST article written by Anette Duensing, M.D.
Gastrointestinal stromal tumors (GISTs) are caused by mutations in the KIT or PDGFRA (platelet-derive growth factor receptor alpha) receptor tyrosine kinase genes and can successfully be treated with imatinib mesylate (Gleevec®). However, despite dramatic response rates and more than 85 percent of the patients initially benefitting from therapy, imatinib treatment is not a cure.
A major problem of imatinib therapy is that a considerable proportion of patients develop resistance to the drug over the course of treatment. A second, interrelated problem of imatinib therapy is the fact that although many patients do achieve tumor stability, complete remissions are rare. In other words, oftentimes even bulky tumor remains under therapy and tumors relapse once imatinib therapy is terminated. These observations are corroborated by scan results during therapy that can show detectable tumor mass in the CT scan although the tumor is responding to imatinib according to a “cold” PET scan. These findings can be interpreted in a way that not all cells have undergone apoptosis in response to imatinib therapy, but instead have significantly reduced their metabolism (which is measured by the PET scan). These remaining cells pose a significant risk to the patient as it is conceivable that they may already contain imatinib resistance mutations that can ultimately contribute to disease progression.
It is therefore imperative to define the exact molecular mechanisms of action of imatinib with the two major questions being (a) how does imatinib lead to GIST cell death (apoptosis) and (b) how can we explain or potentially circumvent incomplete remissions? Answering these questions will not only identify the molecular players that are involved, but also lead to the definition of novel therapeutic targets. Ultimately, our goal is to overcome resistance with an aim towards cure.
Addressing the question of imatinibinduced apoptosis, we have recently identified one of the molecular players: histone H2AX (1). Histones have long been known as mere structural scaffold proteins that help to package the cell’s DNA into its nucleus. They form the socalled nucleosomes, around which the DNA is wrapped. More recently, a plethora of novel functions of histones have been discovered. One major finding was the identification of histone H2AX as a key player in the signaling cascades that are activated after the cellular DNA is damaged (for example, by gammairradiation such as X-rays). We have now discovered yet another function of histone H2AX, which is separate from its role in DNA damage response. Histone H2AX is causatively involved in the induction of GIST cell death after imatinib treatment and can also lead to apoptosis in other cell types that are not treated with imatinib (2). We found that levels of H2AX are massively upregulated in GIST after imatinib, and that most of the H2AX molecules in this context are not part of the nucleosomes, but are free in the cytoplasm or nucleus of the cell. We have shown that the mechanism by which H2AX leads to GIST cell apoptosis involves the inhibition of gene transcription. Importantly, we can now look for alternative ways to target H2AX upregulation as a possible means to circumvent imatinib resistance. (For more detailed information on histones, histone H2AX and its role in imatinib-induced apoptosis please see the December 2007 issue of the LRG newsletter.) Addressing the second question, the problem of incomplete remissions during imatinib treatment, leads us to a fundamental topic in tumor biology and therapy: tumor cell quiescence (3). Quiescent cells are not only unlikely to undergo apoptosis, but they also do not respond to conventional anticancer treatments that target dividing cells. Therefore, tumor cell quiescence is an important problem for cancer therapy in general.
The definition of cellular quiescence is quite simple. Quiescent cells are cells that lack growth/proliferation and that have exited the cell division cycle. Quiescence is a reversible mechanism, which is in contrast to another cellular state called senescence that is irreversible. One of the main molecular regulators of quiescence is the cyclin dependent kinase (CDK) inhibitor p27Kip1, whose levels are high in cells that have entered quiescence (4). P27Kip1 levels on the other hand are regulated by a protein called SKP2. Since the function of SKP2 is to prepare p27Kip1 for degradation, SKP2 levels need to be kept low in quiescent cells. In contrast, dividing cells have high levels of SKP2, which results in low levels of p27 Kip1 (5). The complex molecular events of cell cycle regulation in proliferating and quiescent cells are also depicted in Figure 1.
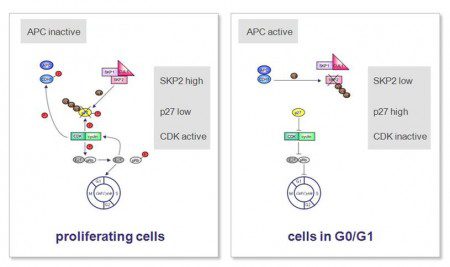 In an attempt to explain the fact of incomplete remissions, we asked whether imatinib is not only capable of inducing GIST cell apoptosis, but could potentially also lead to tumor cell quiescence in GIST cells. To address this question, we treated a GIST cell line model (GIST882) with imatinib and looked for changes in the levels of several known regulators of quiescence, including SKP2 and p27Kip1. We indeed found that levels of SKP2 decreased in a timedependent manner after imatinib treatment as would be expected for cells exiting the cell division cycle. Consequently, because low levels of SKP2 are not sufficient to earmark p27Kip1 for destruction, levels of p27Kip1 increased over this period of time indicating that the cells were entering quiescence as a direct result of imatinib treatment (6).
In an attempt to explain the fact of incomplete remissions, we asked whether imatinib is not only capable of inducing GIST cell apoptosis, but could potentially also lead to tumor cell quiescence in GIST cells. To address this question, we treated a GIST cell line model (GIST882) with imatinib and looked for changes in the levels of several known regulators of quiescence, including SKP2 and p27Kip1. We indeed found that levels of SKP2 decreased in a timedependent manner after imatinib treatment as would be expected for cells exiting the cell division cycle. Consequently, because low levels of SKP2 are not sufficient to earmark p27Kip1 for destruction, levels of p27Kip1 increased over this period of time indicating that the cells were entering quiescence as a direct result of imatinib treatment (6).
We next asked which pathways downstream of KIT are important for entering quiescence after imatinib treatment. It has been shown previously that the socalled PI3K/AKT/mTOR pathway is crucial for GIST cell survival and that a parallel pathway, the MAPK pathway, is less important in this context. Interestingly, we now found that the same is true for the induction of quiescence after imatinib treatment (7).
The next question that we addressed was whether undergoing apoptosis or exiting the cell cycle after imatinib therapy are events that are mutually exclusive. In fact, our experiments showed that nearly all cells that did not die in response to imatinib treatment indeed entered quiescence as indicated by positivity for p27Kip1. These results indicate that imatinib treatment provokes an either- or decision with respect to apoptosis or quiescence. However, it is not clear at this point how this decision is triggered within a cell. We then went one step further and engineered GIST cells to express high amounts of the SKP2 protein. When these cells were treated with imatinib, they were protected from cell cycle exit and kept proliferating. This indicates that the SKP2/ p27Kip1 axis is actively involved in regulating the entry into quiescence during imatinib.
Finally, we wondered whether it would be possible to make use of our findings and translate them into a clinical application. We decided to look at levels of the SKP2 protein in a number of GIST resection specimens and to correlate the results with clinical parameters. We designed a small tissue microarray containing samples from about thirty primary GISTs. When we stained the array for expression of the SKP2 protein, we found that the level of SKP2 positively correlated with the mitotic index, indicating that these two parameters are related to each other. More importantly, we found that the level of SKP2 expression also correlated with an increased risk for recurrence making high SKP2 expression a potential prognostic parameter.
In closing, I would like to introduce you to our current model of the molecular mechanism of imatinib in GIST (Figure 2). Our findings 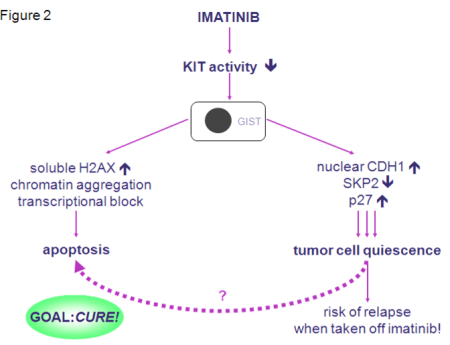 provide evidence that imatinib has several modes of action in GIST. It is not only able to induce apoptosis, but also to directly induce tumor cell quiescence. Therefore, imatinib itself seems to set the stage for future resistance by leading to a pool of cells that are not actively dividing, but that are not dead and may already contain resistance mutations. These cells could ultimately give rise to an imatinibresistant clone. Our findings could also be the explanation for the fact that even patients that do respond to therapy still have bulky disease, which bears the risk of relapse when taken off imatinib. We do not know which of these two pathways prevails in an individual setting. This could be different from patient to patient and from tumor to tumor and could possibly depend on many factors, such as the mutation type as well as others that still need to be defined. Nevertheless, our results indicate that in pursuing our goal to find a cure for GIST we may need to look for pathways that can manipulate quiescent tumor cells to undergo apoptosis.
provide evidence that imatinib has several modes of action in GIST. It is not only able to induce apoptosis, but also to directly induce tumor cell quiescence. Therefore, imatinib itself seems to set the stage for future resistance by leading to a pool of cells that are not actively dividing, but that are not dead and may already contain resistance mutations. These cells could ultimately give rise to an imatinibresistant clone. Our findings could also be the explanation for the fact that even patients that do respond to therapy still have bulky disease, which bears the risk of relapse when taken off imatinib. We do not know which of these two pathways prevails in an individual setting. This could be different from patient to patient and from tumor to tumor and could possibly depend on many factors, such as the mutation type as well as others that still need to be defined. Nevertheless, our results indicate that in pursuing our goal to find a cure for GIST we may need to look for pathways that can manipulate quiescent tumor cells to undergo apoptosis.
References
(1) Liu Y, Tseng M, Perdreau SA, Rossi F, Antonescu C, Besmer P, Fletcher JA, Duensing S, Duensing A. Histone H2AX is a mediator of gastrointestinal stromal tumor (GIST) cell apoptosis following treatment with imatinib mesylate. Cancer Research 2007; 67:2685- 2692.
(2) Liu Y, Parry JA, Chin A, Duensing S, Duensing A. Soluble histone H2AX is induced by DNA replication stress and sensitizes cells to undergo apoptosis. Molecular Cancer 2008; 7:61.
(3) Jackson RC. The problem of the quiescent cancer cell. Advances in Enzyme Regulation 1989; 29:27–46.
(4) Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Müller U, Krek W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nature Cell Biology 1999; 1:207–14.
(5) Bashir T, Pagano M. Don’t skip the G1 phase: how APC/CCdh1 keeps SCFSKP2 in check. Cell Cycle. 2004; 3:850-852.
(6) Liu Y, Perdreau SA, Chatterjee P, Wan L, Kuan, SF, Duensing A. Imatinib mesylate induces quiescence in gastrointestinal stromal tumor (GIST) cells through the CDH1-SKP2- p27Kip1 signaling axis. Cancer Research 2008, 68:9015-23.
(7) Bauer S, Duensing A, Demetri GD, Fletcher JA. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-Kinase/AKT is a crucial survival pathway. Oncogene 2007; 26:7552-7559.