What is GIST?
It is estimated that 3,000-5,000 people develop gastrointestinal stromal tumors (GISTs) in the United States each year (and rates in other countries are similar). GIST, once regarded as a single disease, is now divided into several different forms, based on the presence of specific mutations (genetic changes) in the cancer cells. Most types affect adults, but some affect children, and a few are familial (running in families). Mutations in several genes have been identified as “drivers” of GIST cancers, including the KIT, PDGFRA, SDH, and NF1 genes. Understanding GIST can be challenging.
In some cases, no driver mutation has been identified. These are classified as “wildtype” GIST. As science advances, we expect that mutations driving these wildtype GISTs will be identified. However, regardless of the type of GIST that a patient has, learning as much as possible about the disease is important. As we say, “the wonder drug of the 21st Century is the informed patient”.
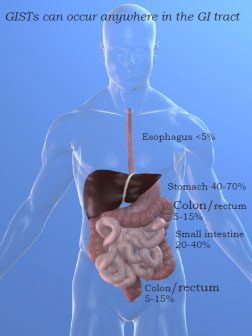
GIST accounts for ~ 1% of cancers of the GI tract.
What type of cancer is GIST?
GISTs belong to a group of cancers called soft tissue sarcomas. Sarcomas are less-common cancers that develop in connective or supporting tissues. Sarcomas are derived from the general class of cells known as “mesenchymal cells”, found in bone, muscle, fat, cartilage, etc. In contrast, most, more common cancers, such as lung, skin, and prostate cancer, develop from epithelial cells which are found on the linings of the body’s many surfaces; such cancers are called carcinomas.
Why does this distinction matter?
Carcinomas and sarcomas behave very differently and are also treated very differently. Since sarcomas are far less common than carcinomas, there are fewer oncologists who specialize in their treatment. See our database of GIST Specialists.
Where do GISTs usually occur?
GISTs may arise anywhere in the digestive tract. The most common sites are the stomach (40-70%), the small intestine (20-40%), and the colon and rectum (5-15%). GISTs can also be found in the esophagus (<5%). (Although very rare, GISTs can develop outside the intestinal tract in the abdominal cavity, these are called eGISTs.)
At the time of diagnosis, most GISTs (80-85%) are limited to a single site (“localized”). However, as the disease progresses, GIST may spread from the original (primary) site to distant locations. If this happens, these secondary tumors are called metastases (or simply, “mets”). It is less common (15-20%) for patients to be metastatic at diagnosis. If GIST tumors do metastasize, they usually travel to the liver, or the peritoneum. Metastases to the lymph-nodes and lungs are rare but do also occur.
It is important to understand that when GIST spreads to the liver, for example, those tumors found on the liver are still GISTs, not liver cancer. GIST metastases in the liver are derived from GIST cells and must be treated like GIST cells. Liver cancer is a completely different cancer that starts in the liver.
Diagnosis of GIST can be difficult – GIST tumors can grow quite large before they are found, and primary tumors often produce few symptoms. GISTs are often spotted during emergency surgery brought about by unexpected perforation of the gastrointestinal tract and consequent bleeding. When GIST tumors are first discovered, the most common symptoms are:
- Vague abdominal discomfort or pain.
- Presence of a palpable abdominal mass.
- Feeling of abdominal fullness.
- Secondary symptoms resulting from tumor bleeding and associated anemia.
Nevertheless, there is no single, typical symptom of GIST. Read more about GIST Diagnosis.
A brief history of GIST
Although the term “GIST” was first used in 1983 (by Mazur and Clark), the 1998 discovery by Hirota and colleagues that GIST tumor cells often have mutations in the KIT gene marked the beginning of a new understanding and reclassification of sarcomas of the gastrointestinal (GI) tract. Prior to the year 2000, GISTs were previously misclassified as other types of soft tissue sarcoma (STS), including tumors of smooth-muscle origin (most commonly leiomyosarcoma, and also leiomyoma or leiomyoblastoma) and those of neural-crest origin (e.g., schwannoma, or nerve sheath tumor).
Most tumors previously diagnosed as Gastrointestinal Autonomic Nerve Tumors (GANTs) are also now classified as GISTs and contain essentially the identical KIT mutations as GIST. What establishes GIST as a separate diagnosis from these other soft tissue sarcomas is not only the description of where the tumor is located, but also the presence or “expression” of the KIT protein (also called CD117). Most GIST patients are also “CD34 positive”, “DOG1 positive” and “desmin negative”. A small percentage of GISTs are KIT-negative.
GIST research is continually changing
GIST cancer research and clinical practice are advancing quickly. We have attempted to make the information presented on this website accurate at the time of posting. Our authors are not medical professionals and the information presented should not be used as a substitute for consultation with a doctor that is experienced with GIST.
No one can take the place of an experienced GIST team of doctors. This may include a Pathologist, Oncologist, and a Surgeon. GIST is such a rare type of cancer that few doctors have experience treating GIST. The hospitals with top sarcoma treatment centers are likely to have more experience with GIST. They have seen many more GIST patients and are more familiar with the unique needs of GIST patients, including treatment options and managing side effects of GIST drugs.
Next Steps:
Science: Biology – Genes and Proteins
Treatments for GIST
Newly Diagnosed Patients
GI Stromal Tumors: A PatholoGIST’s Point of View

This webcast is available in English and Spanish.
9/30/22 ct