The human genome has approximately 30,000 different genes. Each of these genes is contained within the DNA in each cell in the body. When genes are “expressed”, they tell the cell to manufacture specific types of proteins. Some of these proteins are used to communicate with other cells/genes. Cells use “receptors” (and other methods) to listen for the messages (which are often other proteins) of other cells. These receptors are manufactured according to instructions provided by their respective genes within their own cell.
Oncogenes and tumor suppressor genes

How a protein is made from instructions contained in a gene
The most important discovery ever made in basic cancer research is that cancer cells have mutations in specific genes, called “oncogenes” and “tumor suppressor genes”. Harold Varmus and Michael Bishop discovered the first oncogene in 1976 and won the Nobel Prize for the discovery. Many other oncogenes were discovered later, including “KIT”.
Oncogenes are genes that are involved in promoting or regulating cell growth. When mutated, these are the genes that contribute to or cause cancer. A normally functioning (non-mutated) oncogene is called a “proto-oncogene”. When mutated and involved in cancer it is called an “oncogene”. Most proto-oncogenes are components of cell-signaling pathways. Through such pathways, cells can receive a stimulus to undergo mitosis (division) or apoptosis (programmed cell death). Oncogenes and tumor suppressor genes can be compared to the controls of a car. In cancer cells, oncogenes act like a “stuck gas pedal”, and tumor suppressor genes act like “broken brakes”. KIT is the most important oncogene in GIST. Activated KIT (or, in a few cases, a closely-related protein called PDGFR-alpha) is the primary cellular event that causes most GISTs.1 Other oncogenes involved in some GISTs are BRAF (rare) and KRAS, HRAS, and NTRK fusions (very rare).
Tumor suppressor genes are involved in inhibiting cell growth. Mutations that inhibit or inactivate tumor suppressor genes contribute to cancer. Mutations and other defects can result in defects in a tumor suppressor protein called succinate dehydrogenase (SDH). This results in a different type of GIST, called pediatric-like GIST or SDH-deficient GIST.
C-Kit
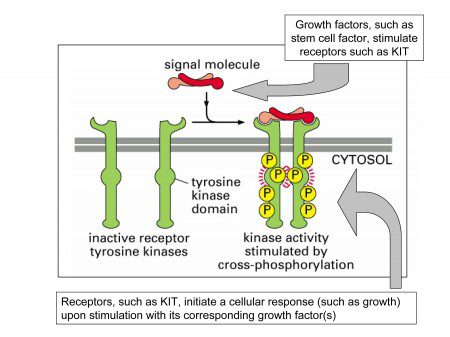
The c-Kit gene contains instructions that tell the cell how to manufacture the KIT receptor. The KIT receptor is a protein in the membrane that envelops the cell. A portion of the receptor is outside of the cell (the extracellular domain), a portion is inside the cell (the intracellular domain), and these two domains are connected by a portion that spans the membrane of the cell (the transmembrane domain).
A “domain” is simply a region of a protein. Just as a house contains several connected rooms, most proteins contain several connected domains. In a house, particular rooms (bathroom, kitchen, dining room) have specific functions.
In a normally functioning cell that expresses c-Kit, another protein called “stem cell factor” binds to the outer portion of the receptor and activates the receptor through a process called phosphorylation. The phosphorylation starts a series of chemical reactions inside the cell. Activation of the KIT receptor causes downstream signaling that results in cell proliferation and survival. (Other names for stem cell factor are steel factor, kit ligand, and mast cell growth factor.)
The c-Kit gene is mutated in approximately 75%-80% of GISTs. When the KIT gene is mutated, the KIT receptor is not assembled correctly. Mutations in KIT result in a change in shape and function of the KIT receptor. Mutations in the KIT gene can cause the KIT receptor to be continuously activated! This is a very important concept. This means that even without the normal signal to divide (stem cell factor), the receptors are still providing a constant stimulus for the GIST tumor cells to divide and survive.
NOTE: KIT, c-Kit and CD117 are all commonly used names for KIT. CD117 or c-Kit is more often used to refer to the stain used for KIT and you are more likely to see this used on a pathology report. C-Kit and KIT are often used interchangeably.
What about the GISTs which apparently do not have mutated c-Kit? But we now know that some of the GISTs that don’t have a mutation in KIT have a mutation in a gene for another protein, PDGFR-alpha, which is very similar to KIT. About 5-8% of all GISTs have a mutation in the PDGFRA gene.
Mutations in the c-kit gene are a very large part of what drives a GIST cancer cell. Because of this fact, KIT is an excellent “drug target”. In other words, we can design drugs that block the signaling of a specific cancer-causing gene/protein, such as c-kit in GIST. A superstar drug in the world of cancer is Gleevec, a drug that inhibits both KIT signaling and PDGFR-Alpha as well as a similar type of protein, BCR-ABL, that causes Chronic Myelogenous Leukemia (CML). Gleevec is approved for both preventative and first-line treatment of GIST. Two other drugs that inhibit KIT (and other proteins) are also approved for use in GIST; Sutent for 2nd line therapy (and for patients who are intolerant to Gleevec) and Stivarga for 3rd line treatment.
The information in genes is divided into different sections called exons and introns. Exons contain protein-coding information and introns do not. Mutations in different exons in the kit gene cause changes in the shape of different parts of the KIT receptor. Mutations in the following exons of the c-kit gene are known to occur in GIST.
Exon 11 is the most commonly mutated exon in GIST. Exon 11 mutations are found in 60% of cases. Mutations in exon 11 generally respond very well to treatment with Gleevec.
Exon 9 mutations are the second most common mutation. Exon 9 mutations are found in 10% of cases. GISTs with exon 9 mutations have a lower response rate to Gleevec therapy when compared to exon 11 mutations, but better than wildtype GIST. They also seem to respond better to higher doses of Gleevec.
Exon 13 and exon 17 mutations are rare in GIST. Because they are so rare, less is known about the response rate to Gleevec. In general, they appear to be responsive to Gleevec, with the exception of the very rare D816V mutation in exon 17.
Some GIST tumor cells do not contain c-Kit or PDGFRA mutations. This may be called wildtype GIST, although, as stated earlier, most of these GISTs are now known to have mutations in other genes. Wildtype GISTs do not respond as well as other types. In general, wildtype GIST encompasses several different subtypes. Advanced mutational testing is available to identify the driver(s) of these tumors.
Important biochemical concept: specific exons usually correspond to specific domains of a protein. That is, exons are like blueprints for particular rooms in a house. This explains why GISTs with mutations in a particular exon, say exon 9, often behave similarly – but differently from GISTs with mutations in another exon – say, exon 11. By “behaving differently” we mean that they may cause primary tumors at different places in the GI tract, and they may respond differently to drugs like Gleevec.
Atypical GIST
In some cases, patients with c-Kit positive GIST may not respond to Gleevec. In particular, certain mutations in the c-Kit gene may prevent the binding of Gleevec to KIT. There are also rare cases in which c-Kit mutations result in the elimination or inactivation of the ligand-binding domain, rendering the tumor c-Kit negative but still responsive to Gleevec. It may be possible in these cases to detect such mutations by mutational testing.
Want more information?
(While these articles are still revelant, there have been significant scientific advances in our
understanding of GIST particularly those without KIT/PDGFRa mutations.)
KIT & PDGFRA Mutations in GIST: A to Z by Dr. Michael Heinrich
Looking for Kinase Mutations in GISTs: How, When and Why? by Dr. Christopher Corless
References
- Michael C. Heinrich, MD, Drian P. Rubin, MD, PhD, B. Jack Longley, MD, and Jonathan A. Fletcher, MD-Biology and Genetic Aspects of Gastrointestinal Stromal Tumors: KIT Activation and Cytogenetic Alterations
- Differential Diagnosis of GIST in the Era of Molecularly Targeted Therapy Rediscovering GIST (Novartis)
9/30/22ct