Click these icons for additional resources for SDH-Deficient GIST patients including fact sheets, specialists, publications, support groups, webinars & videos and more.
Credit: WikiCommons
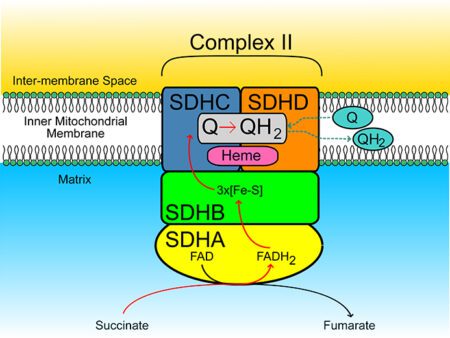
What is SDH?
SDH is a mitochondrial enzyme complex called succinate dehydrogenase. Enzymes act as catalysts that cause a chemical reaction. SDH speeds up the conversion of succinate to fumarate. This conversion is an important part of cell metabolism. The SDH complex plays a critical role in the citric acid cycle known as the Krebs cycle (a cell energy cycle) which drives every cell. (The Krebs cycle is the sequence of reactions by which most living cells generate energy during the process of aerobic or oxygen-based respiration.)
The SDH complex acts as a tumor suppressors – when the complex is functioning it prevent cells from growing and dividing uncontrollably. SDH-deficiency is caused by a loss of function in one of the genes encoding the SDH sub-units (SDHA, SDHB, SDHC and SDHD). A mutation in the SDHx subunits can result in SDH-deficiency. So can an epimutation causing the SDHC gene to not be translated. SDH-deficient GISTs are a unique class of GIST defined by negative immunohistochemical staining (IHC) for succinate dehydrogenase B (SDHB). Both mutant and epimutant forms of SDH deficiency will cause a negative SDHB stain.
In the past, GIST patients who tested negative for KIT/PDGRFA mutant GIST and unresponsive to standard drug treatment protocol for GIST were categorized as wildtype. SDH-deficient GIST, identified in 2011*, has many similarities to KIT/PDGRFA mutant GIST but possesses a profoundly different driving mechanism. It is still called GIST because of the context (as a sarcoma) in which it is found. Genetic studies have identified the mutation/epimutation of additional genes, such as the succinate dehydrogenase (SDH) A, B, C, and D genes. These more recently identified mutations can be classified as SDH-deficient, SDH-epimutant, or SDH-competent (such as BRAF, NF1, NTRK).
*At the 2011 National Institutes of Health (NIH) Pediatric and Wildtype GIST Clinic, pediatric sarcoma specialists studying wildtype GIST patients confirmed the defect in SDH in one of the largest series of young wildtype GIST patients ever studied. Note: ‘Wildtype’ has been a catch-all phrase for “we do not know yet” – identifying those with no mutation found in KIT or PDGFRA. As more mutations are being identified, the wildtype category is becoming obsolete.
Related Syndromes
Patients who test positive for SDHx mutations may be at risk for two related syndromes: Carney Triad and Carney-Stratakis Syndrome.
Carney Triad is a syndromic condition that can include GIST, paragangliomas and pulmonary chondromas. This SDHC epimutation is found mostly in younger patients, and does not appear to be heritable, unlike other SDHx gene mutations. Carney Triad is named after Dr. J. Aidan Carney who first described it in 1977. In Carney Triad, there is impaired SDH function (epigentic SDH inactivation through SDHC hypermethylation), but it is generally not germline. Carney Triad can include GISTs, pulmonary chondromas, paragangliomas (‘complete’ Carney Triad) or simply GISTs and pulmonary chondromas (‘incomplete’ Carney Triad.) The existence of any of these tumors constitutes a diagnosis of CT when age (pediatric) and gender (female) are predominant factors.
Carney-Stratakis Syndrome is an inherited syndrome that affects predominantly young, female patients. Patients with this germline mutation (occurring in any of the SDH subunits) are predisposed to GIST and paraganglioma. If an SDHx mutation is found in the germline, then genetic counseling is indicated, along with mutational screening of first-degree relatives. Patients with Carney-Stratakis Syndrome, caused by a mutation of the SDHB, SDHC, or SDHD, are prone to GISTs, plus other rare tumors, and require regular screening for paragangliomas and pheochromocytoma.
Because of these rare subsets, the need for mutational testing is critical. Targeted medicines that work well for KIT/PDGFRA positive GISTs, in most cases, are not effective for SDH-deficient or SDH-competent mutations. Knowing your mutation will save time spent on a powerful, but ineffective drug, and will hopefully guide you on the path to treatment more suited to your GIST, or if no drug protocol yet exists, to a clinical trial.
SDH and other cancers
Defects in SDH drive other cancers as well, such as paraganglioma, pheochromocytoma, and renal cell carcinoma. Before SDH-deficient GIST was identified in the context of sarcoma, it was studied as a component of a rate syndrome involving neuroendocrine cancer. SDH-deficiency has emerged as a common genetic cause in several cancers.
How Common is SDH-Deficient GIST?
It is estimated that there are approximately 3500-6000 new cases of GIST diagnosed annually in the US. Approximately 10% of these are SDH-deficient.
Diagnosis
The clinical presentation of SDH-deficient GIST is similar to KIT/PDGFRa mutant GIST with one main exception. SDH-deficient GIST occurs almost always exclusively in the stomach. Symptoms of a stomach tumor can include:
- Feeling of fullness
- Nausea
- Loss of appetite
- Loss of weight
- Stomach pain
- Bloody vomit (looks like coffee grounds)
- Constant fatigue/tiredness
- Anemia
- Bloody stools (looks like tar)
- Palpable mass in the upper-right quadrant
Patients, who are initially diagnosed with a stomach mass suspected to be GIST, should undergo an ultrasound guided biopsy prior to the start of any therapy. Immunohistochemistry (IHC) testing of the removed tumor material will determine if it is GIST (CD117 or KIT positive) and if it might be SDHB negative. If the IHC stain for SDHB is negative, the tumor is SDH-deficient and should undergo testing for SDH mutations or epimutations. Patients with SDH-deficient GIST do not require testing for KIT/PDGFRa mutations.
Risk Assessment
This type of GIST behaves differently than KIT/PDGFRA GIST. This disease may present with one or multiple tumors in the stomach (multi-focal). A tumor that is removed surgically may recur, either near the original site (a local recurrence) or at a distant site such as the liver. Local recurrences in the stomach may be a single tumor or multiple stomach tumors. When a tumor recurs at a distant site it is called a metastasis. While all of the above is common both with the KIT/PDGFRA and SDH-deficient GIST, what distinguishes the SDH-deficient it disproportionately affects females and younger people.
Conventional risk stratification parameters appear not to predict metastatic progression of SDH-deficient GISTs as even small, mitotically inactive SDH-deficient GISTs may metastasize, and, as mentioned above, when metastases do occur they may be strikingly indolent, sometimes remaining stable for years or decades1.
Treatment
It is important to find doctors experienced in GIST. This experience comes in two basic types: experts in pediatric cancers and experts in adult GIST cancer. Both have unique perspectives and experiences. The best approach might be when doctors from both fields collaborate with each other.
Surgery
Primary tumor:
Organ preserving surgery is usually the 1st line therapy. Complete gastric resection is to be avoided. It is important to determine the GIST genotype before neoadjuvant treatment or major gastric surgery. Current neoadjuvant therapy (imatinib) is unlikely to work.
Recurrent tumor
Follow-up Surgery to remove blockages and/or to stop bleeding is also appropriate. Salvage or de bulking surgery is not recommended.
Approved Drugs
There are no approved drug therapies specifically for SDH-deficient GIST though there is at least one currently in a clinical trial for the hypermethylated form. The Pediatric & SDH-Deficient GIST Consortium was created to address this issue.
There are drugs approved for KIT/PDGFRa mutant GIST (Sutent, Regorafenib) that have resulted in some reported responses in SDH-deficient GIST. These are case reports and retrospectives and not clinical trials. Response rates have been up to 20%. Currently there are no criteria to predict who will respond.
Next Steps
Contact our Patient Registry Department with any questions about biomarker (mutational) testing, finding an GIST Specialist who is an expert in SDH-deficient GIST, or with questions regarding your treatment or how to proceed after diagnosis.
References
- KIT and PDGFRA Wild-type Gastrointestinal Stromal Tumours (GISTs): ESMO Biomarker Factsheet. Italiano, A.
Oncology PRO Educational Portal for Oncologists.