This article explains hereditary GIST. Familial cancer syndrome is a genetic condition that causes an increased risk for specific types of cancers. Familial cancer syndromes account for only five to ten percent of all cancers.
Most cancer is not inherited. Cancer is common; each year, more than 1.5 million new cancer cases are diagnosed. Many people have relatives who have had cancer, but most of the time this is due to chance or environmental factors (this represents sporadic form of the disease). In a familial cancer syndrome, an inherited genetic mutation causes a person to be at increased risk for cancer and other physical symptoms. There are many different familial cancer syndromes, and each one has a specific set of characteristic cancers and physical symptoms associated with it.
Hereditary forms of GIST arise in the settings of primary familial GIST syndrome or other hereditary syndromes.
Familial GIST syndrome is a rare genetic disorder, showing a high penetrance and representing a small subset of clinical GISTs. To  date, 27 families have been reported. Germline KIT or PDFGRA gene mutations cause most familial cases of GIST. Individuals affected with familial GIST syndrome typically have one normal KIT gene (called wild-type) and one mutant KIT gene. KIT or PDGFRA mutations are similar to those found in sporadic (not familial) form of disease.Familial GIST syndromes show autosomal dominant inheritance, which means that an affected person has a 50 percent chance of passing on the genetic mutation to each of his or her children.
date, 27 families have been reported. Germline KIT or PDFGRA gene mutations cause most familial cases of GIST. Individuals affected with familial GIST syndrome typically have one normal KIT gene (called wild-type) and one mutant KIT gene. KIT or PDGFRA mutations are similar to those found in sporadic (not familial) form of disease.Familial GIST syndromes show autosomal dominant inheritance, which means that an affected person has a 50 percent chance of passing on the genetic mutation to each of his or her children.
Familial GIST syndrome associated with germline KIT mutation is characterized by certain medical and physical features, which are distinct from sporadic GISTs. Patients with this syndrome develop GISTs at a younger age (median – 46 years). The primary tumors are usually multiple in number (3 to >100 tumors) (see Figure 1). The predominant site of tumors development is in the stomach and small intestine, but other gastrointestinal locations are also reported. In addition to GIST predisposition, germline KIT mutations result in other types of gastrointestinal pathology, particularly in disrupted bowel motility, such as difficulty in swallowing (dysphagia), constipation or gastroesophageal reflux (regurgitation). Symptoms associated with gastrointestinal bleeding, such as fatigue, anemia (loss of blood) and melanea (blood in feces) are common and may be the only manifestation of the disease. Additional abnormalities may be present, with a substantial clinical variability. A significant number of familial GIST patients have skin hyperpigmentation, particularly around the mouth, in the perineum, on the face, neck, digits, axillae, groin and knees. Other features that are linked with the dysfunction of melanocytes (pigment producing cells), such as melanocytic moles (colored spots on the outer layer of the skin), lentigines (multiple, tiny, hyperpigmented skin spots), café au lait spots (a pale tan patches of skin) and vitiligo (white patches of skin or hair due to a loss of pigment in the cells) can also be seen. Abnormalities of mast cells, mainly urticaria pigmentosa (a skin rash that is marked by itching and small pale or red swellings) or systemic mastocytosis (abnormal over-production of mast cells) in infancy are less frequently reported.
Germline mutation in the gene encoding PDGFRA has been reported only in three families. In one of these families, a unique combination of multiple fibrous tumors and lipomas of the small intestine and several gastric GISTs was described. This was the second family presented with multiple intestinal tumors. Notably, none of the additional components of familial GIST syndrome (such as hyperpigmentation, dysphagia, or mast cell abnormalities) previously described in germline KIT mutation kindreds were present in the PDGFRA mutation carriers. In two families, all affected family members displayed unusually large hands.
The clinical behavior of familial GIST associated with germline KIT or PDGFRA mutations is the same as sporadic GIST, varying from clinically benign to overtly malignant disease. Imatinib may be effective in the prevention of tumor development as well as in the treatment of familial GISTs. The careful monitoring for the development of tumors is indicated, but the multifocality of the disease suggests that surgical intervention should be avoided in the absence of large tumors or complications.
Other hereditary syndromes associated with an increased risk for GIST are neurofibromatosis type 1 (NF-1), as well as von Recklinghausen’s disease, and Carney– Stratakis syndrome.
Neurofibromatosis type 1 is a condition characterized by changes in skin coloring (pigmentation) and the growth of tumors along nerves in the skin, brain, and other parts of the body. The signs and symptoms of this condition vary widely among affected people. Beginning in early childhood, almost all people with NF-1 have multiple café-au-lait spots, which are flat patches on the skin that are darker than the surrounding area. Freckles in the underarms and groin typically develop later in childhood. Most adults with NF-1 disease develop neurofibromas, which are noncancerous (benign) tumors that are usually located on or just under the skin. These tumors may also occur along nerves elsewhere in the body. People with NF-1 disease also have an increased risk of developing other cancers, including malignant peripheral nerve and brain tumors and leukemia. Neurofibromatosis type 1 is caused by mutations in the NF1 gene. The NF1 gene provides instructions for making a protein called neurofibromin. This protein is produced in many cells, including nerve cells and specialized cells surrounding nerves (Schwann cells). Neurofibromin acts as a tumor suppressor, which means that it keeps cells from growing and dividing too rapidly or in an uncontrolled way. Mutations in the NF1 gene lead to the production of a nonfunctional version of neurofibromin that cannot regulate cell growth and division. As a result, tumors such as neurofibromas can form along nerves throughout the body. Neurofibromatosis type 1 is considered to have an autosomal dominant pattern of inheritance. People with this condition are born with one mutated copy of the NF1 gene in each cell. In about half of cases, the altered gene is inherited from an affected parent. The remaining cases result from new mutations in the NF1 gene and occur in people with no history of the disorder in their family.
Unlike most other autosomal dominant conditions, in which one altered copy of a gene in each cell is sufficient to cause the disorder, two copies of the NF1 gene must be altered to trigger tumor formation in neurofibromatosis type 1. A mutation in the second copy of the NF1 gene occurs during a person’s lifetime in specialized cells, such as cells surrounding nerves, interstitial cells of Cajal (ICC, precursor GIST cells) or blood cells. Almost everyone who is born with one NF1 mutation acquires a second mutation in many Schwann cells and develops neurofibromas. However, the risk of developing GISTs is low (only 7%, based on a single Swedish study)2. GISTs in NF1 patients tend to be multiple and are located predominantly within the small intestine. Abdominal pain, bowel obstruction and massive gastrointestinal bleeding are the most common presenting clinical manifestations. Although they may fall into any GIST risk category, NF1-associated GISTs usually show low cell proliferation (growth) indicators and they rarely metastasize. Vast majority of GISTs associated with NF-1 do not carry KIT or PDGFRA mutations, and for this reason treatment with tyrosine kinase inhibitor drugs such as imatinib might be less effective in NF-1 GIST patients.
In a subset of patients with gastric GISTs the lesions are associated with paragangliomas (PGLs). The condition referred to as the dyad of “paraganglioma and gastric stromal sarcoma” or the “Carney–Stratakis syndrome” is transmitted as an autosomaldominant trait with incomplete penentrance (not all carriers of the mutant gene develop tumors). The patients present tumors at a young age (median age 19 years). GISTs are multifocal and paragangliomas multicentric, supporting a genetic link between the two lesions. PGLs are neuroendocrine tumors that may secrete catecholamines. They occur most frequently in the head, neck, adrenal and extra-adrenal sympathetic ganglia. Once the diagnosis is made, patients must be followed carefully in order to detect new lesions as early as possible, since both, GISTs and PGLs, have malignant potential. A functioning paraganglioma may manifest itself by sympathetic effects such as hypertension, increased perspiration, and/or facial flushing. A 131I-MIBG scan is useful for diagnosis, as this agent localizes to catecholamine-producing tissues.
GIST patients with Carney–Stratakis syndrome do not carry the KIT or PDGFRA gene mutations. The underlying hereditary defect of the Carney– Stratakis syndrome has been elucidated recently by identification of germline “loss-of function” mutations in the succinate dehydrogenase subunit B (SDHB), C (SDHC) or D (SDHD) genes in affected patients. The identified mutations had been described before in sporadic or familial paragangliomas. Notably, the abdominal paragangliomas associated with GISTs are uniquely correlated with SDHC mutations. The absence of KIT or PDGFRA somatic mutations and inactivation of one of the succinate dehydrogenase subunits in GISTs/PGLs from patients with the dyad suggests that a deficient mitochondrial tumor suppressor gene pathway is responsible for tumor formation and not constitutively active tyrosine kinases. The role of imatinib as a therapeutic or preventive intervention in Carney– Stratakis syndrome patients remains to be defined.
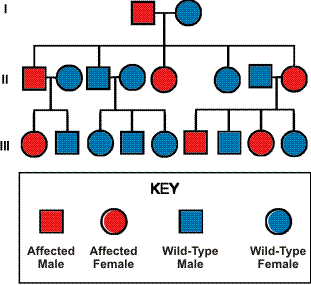
Inheritance pattern of familial GIST (autosomal dominant).
(Adapted from Wikipedia).
Below is a list of “clues” in a family tree that make a familial GIST syndrome more suspicious:
a. GIST diagnosed more than once in the same person (more than one primary tumor, not a cancer recurrence).
b. GIST diagnosed at an earlier age than usual (before age 50)
c. Two or more close relatives with GIST (on the same side of the family)
d. One person in the family who has had a GIST and another cancer as well
e. One person in the family with GIST who also has a personal or family history of unusual skin findings, multiple moles, or NF1.
The most important step in determining if a family has a familial cancer syndrome is gathering an accurate family history. For a person who has had cancer, the type of cancer and age at diagnosis should be listed for each cancer. It may be necessary to obtain medical records to confirm what type of cancer a person had since family members may not always be aware of specific information. Birth defects, unusual skin findings, benign tumors, and special screening tests (such as colonoscopy to look for colon polyps) should also be noted. Many hospitals have a “familial cancer unit,” which is a team of health professionals with expertise in familial cancer syndromes. Geneticists, genetic counselors, oncologists, and social workers assist individuals and families by providing risk assessment, support, screening and prevention recommendations, and genetic testing options. When a person is diagnosed with a familial cancer syndrome, relatives should be examined for signs of the syndrome. Sometimes a person identified as having a familial cancer syndrome is the first person in the family to be affected. That person is able to pass the condition on to his or her children, but the parents and siblings are not affected. Depending on the syndrome, genetics professionals can determine who in the family is at risk. There are special issues surrounding genetic testing that should be discussed; for example, what age should the test be done (important for children)? How would the results change medical management? Does insurance cover the test? Is prenatal diagnosis indicated? How will the information affect the family? Health professionals familiar with familial cancer syndromes keep up to date with advances in cancer genetics, and work with families to discuss the risks, benefits and limitations of genetic testing.
Points to remember:
• A person only needs to inherit one copy of the changed gene in order to be affected by the condition (50% chance). These outcomes occur randomly. They remain the same in every pregnancy and are the same for boys and girls.
• A changed gene cannot be corrected – it is present for life.
• A changed gene is not something that can be caught from other people. They can still be a blood donor, for example.
• People often feel guilty about a genetic condition which runs in the family. It is important to remember that it is no one’s fault and no one has done anything to cause it to happen.
1. Antonescu CR. Gastrointestinal stromal tumor (GIST) pathogenesis, familial GIST, and animal models. Semin Diagn Pathol. 2006;23 (2):63-9.
2. Cummings, S. “The Genetic Testing Process: How Much Counseling Is Needed?” Journal of Clinical Oncology 18, Supplement (November 1, 2000): 60–4.
3. Li FP, Fletcher JA, Heinrich MC, Garber JE, Sallan SE, Curiel-Lewandrowski C, Duensing A, van de Rijn M, Schnipper LE, Demetri GD. Familial gastrointestinal stromal tumor syndrome: phenotypic and molecular features in a kindred. J. Clin. Oncol. 2005;23, 2735-2743.
4. Maertens O, Prenen H, Debiec-Rychter M, Wozniak A, Sciot R, Pauwels P, De Wever I, Vermeesch JR, de Raedt T, De Paepe A, Speleman F, van Oosterom A, Messiaen L, Legius E. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum. Mol. Genet. 2006, 15,1015-1023.
5. Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos SS, Muchow MM, Boikos SA, Ferrando B, Pacak K, Assie G, Baudin E, Chompret A, Ellison JW, Briere J-J, Rustin P, Gimenez-Roqueplo A-P, Eng C, Carney JA, Stratakis CA. Clinical and molecular genetics of patients with the Carney–Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur. J. Hum. Genet. 2008;16, 79-88.
6. Robson ME, Glogowski E, Sommer G, Antonescu CR, Nafa K, Maki RG, Ellis N, Besmer P, Brennan M, Offit K. Pleomorphic Characteristics of a Germ-Line KIT Mutation in a Large Kindred with Gastrointestinal Stromal Tumors, Hyperpigmentation, and Dysphagia. Clin. Cancer Res. 2004;10, 1250-1254.
If you want to learn more about Familial GIST:
-
Visit the Dana Farber website for more information on Project FLAG
-
Visit our webcast archive to watch the Familial GIST webcast
-
Look out in the coming month for a new LRG Familial GIST pamphlet!