Prior to 2000, surgery was the only known effective treatment for advanced GIST. Notably, conventional sarcoma chemotherapy yielded response rates of less then 5 percent and very short time intervals before disease progression/treatment failure. In retrospect, these treatments were probably more harmful than helpful.
The GIST world changed dramatically with the discovery of KIT activating mutations in 1998. This discovery set the framework for clinical trials of imatinib (Gleevec) that began in 2000. Over the past 13 years, the use of KIT-targeted therapy has revolutionized the treatment of advanced GIST with high response rates, tolerable side effects, impressive palliative effects on tumor-associated symptoms like abdominal distention/pain, durable disease control, and improved overall survival.
Currently, overall survival is estimated to be in the range of five to seven years for patients with newly diagnosed metastatic GIST. In addition, a sizable minority of patients treated for metastatic GIST will experience durable disease control lasting more than a decade.
However, the majority of metastatic GIST patients treated with imatinib will experience disease progression within three years of starting treatment. Molecular analyses of drug-resistant tumors have identified acquired (secondary) kinase mutations as the cause of drug resistance in most cases. Imatinib, and similar drugs, need to bind to KIT in order to block its activity.
Secondary mutations prevent imatinib (or other drugs) from binding correctly (or even at all). This can be thought of in terms of a lock and key model, where the lock is the KIT protein and the key represents imatinib. If you change the lock, the key will no longer work. Secondary mutations are the cause of most cases of drug-resistant KIT exon-11 mutant GIST. The mechanisms of resistance in patients whose GIST lack KIT or PDGFRA mutations are more complicated and varied, due to the fact that this subset of GIST is actually composed of at least 10 different types of tumors.
The success of front-line treatment with imatinib, coupled with the discovery that many imatinib-resistant GIST are still “addicted” to KIT, lead to clinical studies of other KIT inhibitors. As of 2013, sunitinib and regorafenib have been clinically validated and approved by health regulatory authorities (e.g. the U.S. Food and Drug Administration) for the treatment of drug-resistant metastatic GIST.
Once the initial results with imatinib were known, some investigators wondered whether or not continuous treatment was needed to maintain disease control. Historically, patients with advanced cancer have been treated intermittently rather than continuously. This was due in part to the toxicity of continuous chemotherapy treatment, and also because there was no evidence that continuous treatment was better than intermittent treatment. The prevailing cancer treatment paradigm prior to imatinib was chemotherapy treatment to cure patients with minimal disease after surgery (e.g. colon or breast cancer) or to cure patients with highly chemotherapy-sensitive diseases like Hodgkin lymphoma. For most patients with metastatic cancer, chemotherapy was used to halt disease progression and/or extend survival and was given when needed but not continuously.
To determine whether continuous imatinib was needed for optimal GIST disease control, investigators from the French Sarcoma Group randomized long-term responders (1, 3, or 5 years) to either continue imatinib treatment or to stop and undergo close surveillance. The primary endpoint of this study was to determine the effect of stopping vs. continuing treatment on disease control. The results for patients randomized after three years of imatinib treatment are shown in figure 1.(1) As is clearly seen by the separation of the curves, almost all patients who stopped treatment had disease growth within one to two years. In contrast, patients who continued treatment had a much slower rate of disease growth. These data support the need for continuous imatinib treatment and indicate that long-term disease control does not mean that we have eliminated all GIST cells, as tumors start growing within months of stopping treatment.
The two curves from this study identify two different tumor states: 1) the rapid relapse (tumor progression) of the patients who stop treatment is due to persistent disease; and 2) the slow loss of tumor control over time for the patients who continue imatinib-treatment is due to the emergence of resistant disease.
As noted above, drug resistance can arise from multiple mechanisms, most commonly from the development of secondary KIT mutations. In contrast, persistent disease reflects a population of cells whose growth is inhibited by imatinib, leading to some form of cellular “hibernation.” These hibernating cells can survive continuous imatinib treatment but, over time, these same cells may develop additional mutations and evolve into drug resistant tumors.
Although the mechanisms underlying these two cellular states are different, they share one central shared feature: in both cases, the clinical attempt to block KIT signaling is insufficient to induce cellular death. In the case of persistent disease, even near-total inhibition of KIT does not cause cellular death, due to the existence of other survival mechanisms. In the case of resistant disease, minimal or sub-optimal KIT inhibition by available drugs like imatinib, sunitinib, or regorafenib is insufficient to cause tumor cell death and in some cases does not even slow the growth of these tumors.
Theoretically, more potent KIT inhibitors with an enhanced spectrum of activity against different resistance mutations would improve upon current treatments, especially in the case of resistant disease. However, even this approach will not solve the problem of persistent disease.
As previously discussed in this forum, many GIST scientists and clinicians feel that a joint solution to treating persistent, as well as resistant, disease is the use of combination treatments. Such an approach would exploit the known benefits of inhibiting KIT enzyme activity and add in the effects of blocking a separate critical survival pathway. Theoretically, some form of combination therapy might eradicate persistent disease, thereby leading to a cure. Finding the right combination to treat GIST has become the goal of many GIST scientists, including all the members of the Life Raft Group Research Team.
To identify targets to include in a combination treatment, one can use either a biased or an unbiased target identification approach. In the biased approach, we use existing knowledge to suggest new drug targets. This approach is exemplified by Dr. Rubin’s recent commentary in the June 2013 newsletter. Notably, signaling from KIT tends to be transmitted via two main pathways known as PI3K and MAPK. Both pathways are known to be important to GIST growth and survival and both pathways are activated by KIT signaling. These two pathways are analogous to two main highways that lead out of a large city. If there is a traffic jam on one highway, one can still leave the city by using the other. In the case of GIST, cells manage to figure out a way to restore signaling through one or both pathways.
In his commentary, Dr. Rubin presented evidence that a triple combination of a KIT inhibitor + MAPK pathway inhibitor + a PI3K pathway inhibitor was much more effective than single or dual drug treatment. This result highlights the advantages of using a biased approach: scientists can focus on what is already known about GIST biology and immediately proceed to laboratory studies of combination treatments. However, this approach also has limitations: 1) we have not discovered everything we need to know about GIST biology and may not have identified the critical or optimal pathways to drug; and 2) some pathways that we know are important to GIST biology may not provide good drug targets (e.g. transcription factors like ETV1).
In contrast, the unbiased screening approach utilizes one or more technologies to identify targets, even those that have never been identified as important to GIST biology, or any cancer for that matter. A number of members of the LRG Research Team are using large scale screens to identify novel pathways and targets in GIST.
Dr. Jonathan Fletcher’s group has been using RNA interference (a method of blocking protein function) to systematically inhibit 11,194 different proteins in GIST cells–one protein at a time. By combining RNA interference with imatinib treatment, Dr. Fletcher’s group has identified several critical and novel pathways. Their initial results validating CDC37 as a treatment target were recently published by Dr. Marino-Enriquez et al.(2) Their results highlight the main strength of an unbiased screening approach: you can find targets that were not previously known to be important to cancer cells.
Dr. Anette Duensing’s laboratory has been using a related but technically distinct screening approach to identify pathways that could be targeted in GIST in conjunction with KIT inhibitors.
In addition to the work described above, Dr. Brian Rubin also has been conducting screening studies using a chemical library of existing cancer drugs (including kinase inhibitors) to try and identify novel combination treatment approaches. By testing many drugs and combinations, we may find a better treatment.
Over the past several years, my laboratory also has been focused on trying to develop combination treatment strategies for GIST. This work has been conducted in collaboration with Dr. Chris Corless, the above LRG investigators, as well as Drs. Debiec-Rychter and Bauer (both members of the LRG Research Team). One of our approaches has been to sequence all the genes in drug-resistant GIST to find which pathways are commonly mutated in addition to the known KIT mutations in these tumors. These types of experiments produce terabytes of data and take many months to analyze. The datasets from these experiments are too large to analyze on a personal computer and must be stored and analyzed in “the cloud.” The analysis of several dozen sequenced tumors is still ongoing.
More recently, my group started using a novel methodology to identify pathways important to GIST biology. In these experiments, cells are treated with KIT inhibitors and we measure changes in gene expression using an approach known as RNA-Seq. By identifying pathways that are modulated by KIT inhibitors, we hope to find novel treatment strategies.
In our preliminary work, we have identified several novel pathways for which existing drugs or research grade inhibitors already exist. Using these drugs/compounds as research tools, we hope to find new combination treatments that can be further tested in the laboratory and hopefully advanced to clinical studies. Our preliminary results are promising and suggest that this approach is likely to identify a number of targetable pathways. In some cases, drugs to target these pathways already exist, allowing us to more rapidly advance promising combination treatments to clinical studies.
As outlined above, I believe that combination treatments are the key to finding more effective and, hopefully, curative GIST treatments. Your LRG Research Team is working hard to identify these combinations. Due to the close collaboration of our group, new findings are freely shared, leading to more rapid progress. Our common objective is to find a cure for GIST and we believe that working as a team is the best way to achieve that goal. Hopefully, our work on finding new combination treatments will bring us closer to a cure for GIST.
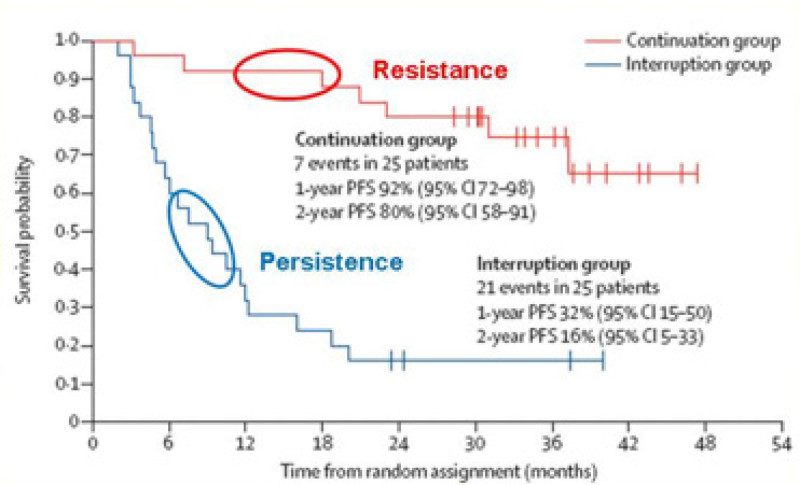
Figure 1. Results from the BFR14 randomized study of interrupted vs. continuation treatment. Data are shown for patients randomized after three years of successful imatinib treatment (i.e. only those patients who were still responding to imatinib). The blue curve represents outcomes after halting treatment. At one year, only 32% of patients had not experienced detectable tumor growth. The red curve is data from patients who continued their imatinib treatment. At the one year time point, more than 90% of patients continued to respond to imatinib (i.e. no tumor growth). As discussed in the text, patients who continue treatment and have subsequent tumor growth have resistant disease. In contrast, persistent disease is responsible for the growth of tumors in patients who stop treatment. Reference: (1)
Reference List
(1) le Cesne A, Ray-Coquard I, Bui BN, Adenis A, Rios M, Bertucci F, et al. Discontinuation of imatinib in patients with advanced gastrointestinal stromal tumours after 3 years of treatment: an open-label multicentre randomised phase 3 trial. Lancet Oncol 2010;11:942-9.
(2) Marino-Enriquez A, Ou WB, Cowley G, Luo B, Jonker AH, Mayeda M, et al. Genome-wide functional screening identifies CDC37 as a crucial HSP90-cofactor for KIT oncogenic expression in gastrointestinal stromal tumors. Oncogene 2013.
