By Tamas Ordog, LRG Research Team, Martin Zörnig and Yujiro Hayashi

Dr. Tamas Ordog
Gastrointestinal stromal tumors (GIST) represent a substantial proportion of human bone and soft tissue sarcomas.1 GIST are thought to share origins with interstitial cells of Cajal (ICC),2-5 a regulatory cell type within the gut musculature.6 ICC and GIST also share several key characteristics including expression of the receptor tyrosine kinase (RTK) KIT,2,3 the calcium-activated chloride channel ANO1 (TMEM16A, DOG1),7,8 protein kinase C-theta,9 and the transcription factor ETV1.10 In contrast, while most GIST and about one-half of ICC precursors express platelet-derived growth factor alpha (PDGFRA), PDGFRA can only be detected in approximately four percent of ICC,5,11 with most gastrointestinal PDGFRA expression occurring in KIT-negative interstitial cells distinct from ICC. However, the role of these cells in GIST oncogenesis remains unclear.12
The majority of GIST arise from mutations in either KIT (75-80%)3 or PDGFRA (<10%).13 The remaining 10-15 percent may contain driver mutations in BRAF, HRAS, NRAS or NF1.14 These tumors are morphologically and clinically indistinguishable from RTK-mutant GIST including expression and activation of KIT. A small subset of adult tumors and the majority of pediatric GIST display unique features including predilection toward females, predominant gastric origin, and epithelioid morphology. This class shows increased expression and activation of insulin-like growth factor 1 receptor (IGF1R)15 and loss of mitochondrial succinate dehydrogenase complex subunit B protein (SDHB), which may arise from several different causes.15 SDHB loss, in turn, leads to aberrant gene expression16 and signaling via mechanisms normally activated by reduced O2 levels.14
The standard of care for patients with a primary localized GIST is surgery. However, approximately 40 percent of patients develop tumor recurrence within five years. Front-line treatment with the KIT/PDGFRA inhibitor imatinib can achieve disease control in 70-85 percent of patients with KIT+ advanced GIST and a median progression-free survival of >5 years.14 Resistance developing after an initial benefit is mainly due to acquired, drug-resistant mutations.17 Unfortunately, resistance mutations show considerable heterogeneity, and, therefore, even second- and third-line drugs have only moderately increased median progression-free survival.18
In patients that respond to imatinib, substantial reduction in tumor size occurs. However, RTK inhibitors fail to eradicate GIST cells in 95-97 percent of patients.14 Although the surviving cells appear non-proliferating, this state is reversible, necessitating life-long treatment. The significance of GIST persistence is that it sets the stage for secondary, drug resistant mutations.18 Therefore, it can be argued that targeting disease persistence should take precedence over the development of additional pharmacological agents against secondary mutations.
GIST persistence during RTK inhibitor therapy could result from “escape” mechanisms expressed by the tumor cells. Alternatively, a pre-existing subset of cells not dependent on oncogenic RTK signaling due, e.g., to lack of significant expression of the mutant receptor could survive the treatment. In seven studies that investigated KIT expression in patients that underwent imatinib or sunitib treatment prior to surgery, 18 of 148 samples lacked KIT expression and further samples expressed low KIT, with the remainder showing no obvious change (reviewed in ref.19 and see ref.11). Thus, both mechanisms may contribute to GIST persistence. Typically, the cells expressing little or no KIT (KITlow/negative) had epithelioid morphology.19 Previously, we described a rare KITlow/negative cell type with epithelioid morphology in mice and demonstrated their ability to self-renew and differentiate into ICC both in vitro and in vivo, signifying their role as ICC stem cells (ICC-SC).4,5,20 Transformed ICC-SC gave rise to GIST-like tumors containing both epithelioid, KITlow and spindle-shaped, KIT+ cells.5 Importantly, both normal and transformed ICC-SC showed low sensitivity to imatinib. These findings are consistent with a GIST model wherein a small number of mutated ICC-SC gives rise to KIT+ cells representing the bulk of the tumors (Figure 1). Whereas RTK inhibition can keep KIT+ GIST cells under control, it may not eradicate the inherently imatinib-resistant KITlow/negative stem cell pool, from which the tumor is reestablished following the cessation of therapy. Acquisition of an imatinib-resistant mutation by the surviving precursors would again permit their differentiation into KIT+ cells and uncontrolled GIST growth.5 It follows that stimulation of KIT expression in the surviving KITlow/negative GIST precursors before the emergence of drug-resistant mutations could potentially restore these cells’ sensitivity to imatinib. Although this model bears remarkable similarities to the model proposed to underlie disease persistence in chronic myeloid leukemia,21 its applicability to human GIST remains to be established.
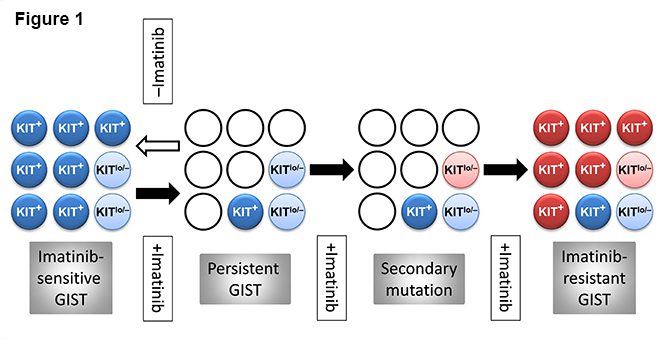
Figure 1. Hypothetical stem cell model of GIST persistence and acquired therapy resistance. Light blue circles: KIT-independent precursors carrying imatinib-sensitive KIT mutation but expressing very little or no KIT protein (KITlow/–). Dark blue circles: KIT+ cells arising from the KITlow/– cells carrying imatinib-sensitive KIT mutation. Open circles: dead cells. Pink circles: KITlow/–, KIT-independent precursors with acquired secondary imatinib-resistant mutation. Red circles: KIT+ cells differentiated from the KITlow/– precursors with secondary imatinib-resistant mutation. Filled arrow: imatinib treatment. Open arrow: cessation of imatinib treatment.
In GIST cells dependent on imatinib-sensitive mutations, disease persistence may reflect incomplete apoptosis (a form of cell death) in response to RTK inhibition.22 GIST cells may escape apoptosis by upregulating macroautophagy (self-digestion of cellular components),23 withdrawal from the cell cycle,24 or entering a state of quiescence.25 Importantly, these mechanisms could be blocked experimentally by antimalarial agents or inhibition of the protein kinase DYRK1A.23-25 Thus, GIST could be sensitized to RTK inhibition-induced apoptosis by pharmacological inhibition of various escape mechanisms or stimulation of differentiation of KITlow/negative precursors. However, the effects of these interventions may be limited by loss in most GIST of FAM96A, a regulator of cellular iron homeostasis which we recently found to have important role in apoptosis.26
GIST cells not dependent on constitutively active RTK signaling must draw on alternative pathways for survival. Pharmacological targeting of these mechanisms may provide additional means to eliminate cells causing disease persistence and also to treat SDHB-deficient GIST, which respond poorly to imatinib and related drugs.15 IGF1R is variably expressed and activated in several GIST subtypes and most robustly in pediatric GIST,15 possibly indicating their relationship to ICC-SC,15 which, unlike mature ICC, strongly express this gene.4 IGF1R inhibition led to cytotoxicity in KITnegative mutant cells,15 and is currently under investigation in a phase II clinical trial in GIST lacking KIT, PDGFRA or BRAF mutations (NCT01560260).
IGF1R may also promote GIST survival by stimulating the expression of stem cell factor, the ligand for KIT, by cells of the ICC/GIST microenvironment.27 This indirect effect requires expression of a wild-type KIT allele (an allele is one of a pair of genes that appear at a particular location on a particular chromosome and control the same characteristic, which is common in GIST). Interestingly, ligand-driven KIT activation may remain active in tumors treated with imatinib due to the preferential targeting of mutant receptors by this drug.28 Recently, we demonstrated a similar role for ligand-dependent activation of wild-type PDGFRA co-expressed with mutant (including imatinib-resistant) KIT.11
In GIST that became KITnegative by long-term exposure to imatinib, transition from spindle-shaped to epithelioid morphology has been shown to be accompanied by overexpression of the RTKs AXL and MET.29 Furthermore, epidermal growth factor receptor (EGFR) expression and activation has been reported in GIST lacking KIT or PDGFRA mutations.30 EGFR expression and activation appears to be common in both imatinib-treated and untreated GIST along with the expression of several EGFR ligands.29,31 Together, these results indicate that activation of alternative RTK pathways by ligands released in the tumor microenvironment may be common in GIST including tumors not dependent on KIT/PDGFRA signaling.
In conclusion, disease persistence in GIST involves multiple mechanisms including activation of signaling pathways triggering the cells’ exit from the cell cycle, autophagy, loss of pro-apoptotic proteins, downregulation of KIT/PDGFRA expression or selection of GIST stem cells that do not depend on KIT/PDGFRA signaling for survival due to expression of alternative receptor tyrosine kinases (Figure 2). In view of the molecular diversity of GIST exposed to long-term imatinib, eradication of residual tumor cells and curing GIST will likely require individualized combinations of several approaches tailored to the tumors’ genotype and phenotype.
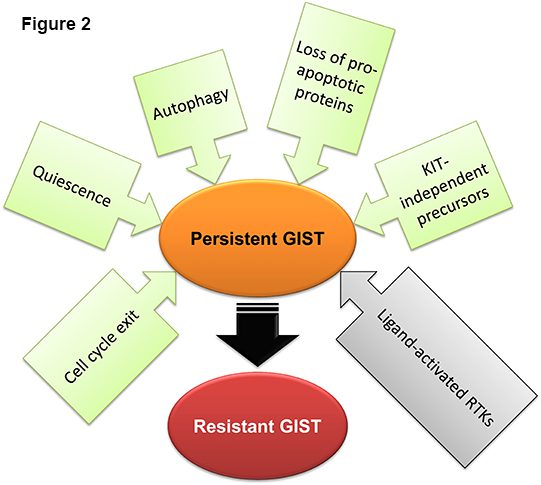
Figure 2. Mechanisms of disease persistence in GIST. Green and gray boxes signify cell-autonomous and microenvironmental mechanisms, respectively.
References
For full references, please view the original article cited here: bit.ly/NLOct2015Ordog
Abridged and modified from the following article: Ordog T, Zörnig M, Hayashi Y. Targeting Disease Persistence in Gastrointestinal Stromal Tumors. Stem Cells Transl Med. 2015 Jul;4(7):702-7. doi:10.5966/sctm.2014-0298. Epub 2015 May 1. PubMed PMID: 25934947; PubMed Central PMCID: PMC4479627. ©AlphaMed Press, 2015.
